Cu toate acestea, toate fenilcetonurie boli ereditare, singurul care poate fi complet neutralizat. Astăzi, un copil născut cu semne de fenilcetonurie, se pot dezvolta perfect sănătoși. creierul copilului Protect este posibil printr-o dietă specială, pe care vom descrie mai jos.
În diferite țări, frecvența bolii este diferită uneori. Rusul născut un copil bolnav la 10 000. În unele părți ale Regatului Unit, această cifră este de două ori mai mare - 1: 5000. Copiii din Africa cu greu suferă de fenilcetonurie. Dintre pacienții cu numărul de fete este aproape de două ori numărul de băieți.
mecanisme de dezvoltare a bolii
Boala este moștenită numai atunci când ambii părinți dau copilului o tendinta de a bolii, și, prin urmare, destul de rare. Doi la suta din oameni au genei alterate responsabile pentru boala. Acest om este complet sănătos. Dar când un bărbat și o femeie, purtătorii genei mutante, se căsătorească și să decidă să aibă copii, probabilitatea ca copiii vor avea de suferit de fenilcetonurie, este de 25%. Iar posibilitatea ca copiii vor fi purtători ai genei anormale PKU, dar ele rămân relativ sănătoși, este de 50%.Cauza acestei boli este asociată cu faptul că o enzimă nu este produs în ficat uman - fenilalaninei 4-hidroxilaza. El este responsabil pentru conversia fenilalanina la tirozină. Ultima parte a pigmentul melanina, enzimele și hormonii necesari pentru functionarea normala a corpului.
Când PKU fenilalanină, rezultând în schimb moduri adverse, convertite în substanță, care ar trebui să fie în organism: și acidul fenilpiruvic fenillactic și feniletilamina ortofenilatsetat. Acești compuși se acumulează în sânge și au o acțiune complexă:
- viola procesele metabolismului lipidic în creier
- induce deficite de neurotransmitator care transmit impulsuri nervoase între celulele sistemului nervos
- au un efect toxic, otrăvire a creierului
simptome de fenilcetonurie
Copiii cu PKU se nasc absolut sănătoși. Prin urmare, dacă în primele zile de viață pentru a identifica boala și să rămânem la dieta, este posibil pentru a preveni distrugerea creierului copilului. În același timp, nu există simptome ale bolii. Copilul crește și se dezvoltă, precum și colegii lui.În cazul în care momentul în care este pierdut, iar copilul mănâncă alimente bogate în proteine, cu fenilalanină, ei încep să prezinte simptome ale sistemului nervos central. Inițial se schimbă la pacienții cu fenilcetonurie sunt nesemnificative. Ele sunt dificil de a vedea chiar și un medic pediatru cu experiență. Această slăbiciune și de anxietate. Copilul zâmbește și se mută mici.
După șase luni de întârziere devine mai evident. Copil slab receptiv la ceea ce se întâmplă, nu știu mama, ea nu încerca să se așeze și să se rostogolească. Fenilalanina și derivații săi sunt excretați în urină și sudoare. Ele provoacă un anumit „mouse“ sau mirosul de mucegai.
copil de ani nu știe cum să-și exprime vocea emoțiilor și sentimentelor lor, este de expresii faciale inexpresiv, nu înțelege părinții.
La vârsta de trei ani și mai mari simptome de fenilcetonurie sunt în creștere. La copii, există iritabilitate, oboseală, tulburări de comportament, tulburări psihotice, retard mintal. Dacă nu se ocupă cu tratamentul de fenilcetonurie, starea pacientului se va deteriora.
Diagnosticul de fenilcetonurie
În acest caz, în cazul în care se suspectează că unul sau ambii părinți sunt purtători ai genei fenilcetonurie, care pot fi determinate în federale centre medicale genetice. Pentru a stabili acest fapt efectuat examinarea genetică.Până în prezent, toate nou-nascuti sunt testati pentru prezența unor cantități mari de fenilcetonurie. În Rusia această întrebare reglementează ordinul Ministerului Sănătății al Federației Ruse №316 din 30.12.1993 Procedura se numește screening-ul neonatal și este un mod eficient de a identifica cele mai comune boli ereditare printre ei, și fenilcetonurie.
screening-ul nou-născutului este o metodă simplă și fiabilă de diagnostic. În spital, fiecare copil să ia câteva picături de sânge de la tocuri periferice. Acest lucru se face pe stomacul gol, la trei ore după hrănire. În analiza sugari pe termen lung să ia în a patra zi de viață, și prematură în a șaptea. Pentru acei sugari care au fost născuți nu în spitale, este important să se ia testul pentru primele trei săptămâni.
Sângele este aplicat într-o formă specială de testare, care este apoi trimis la un laborator pentru cercetare genetica. Acolo pe tot parcursul zilei a efectuat un test de sânge pe conținutul de aminoacizi în ea - fenilalanină. Rezultatele testelor sunt înregistrate în schimbul cardului copil ștampilate „pentru fenilcetonurie și VG examinat.“
În cazul în care analiza detectează o modificare a genei, părinții și copilul sunt invitați la Centrul de Genetica Medicala pentru examinare. Pentru a confirma sau infirma diagnosticul de cercetare suplimentare sunt atribuite:
- în loc de sânge uscat
- ser
- test de sudoare
- coprogram
- ADN-ul diagnostic
tratamentul fenilcetonurie
Astăzi, în țara noastră, singura metodă eficientă de tratament este terapia de dieta. Dezvoltarea de droguri, care va controla nivelul de fenilalanină în sânge fara dieta. În acest sens, există un progres semnificativ, dar vânzarea de astfel de medicamente par să nu mai devreme decât în 5-7 ani.În mod constant lucrăm din greu pentru a găsi noi mijloace și metode de combatere a bolii.
- O direcție promițătoare este utilizarea unui fenilalaninliazy enzimă de plante, care va scinda excesul de fenilalanină în organism.
- Oamenii de știință au speranțe mari pentru terapia genica folosind un factor viral, care va vindeca gena bolnav și complet a scăpa de această problemă.
- Practicat prin administrarea genei hidroxilaza fenilalanina direct în celulele afectate ale ficatului.
Unele forme de fenilketonurie tetrahydrobiopterin tratabile, care este o parte a enzimei lipsă fenilalanină 4-hidroxilaza. Formele atipice de PKU nu este tratat cu dieta și necesită un aport regulat teteragidrobiopterina sau înlocuitorii acestuia.
Mese pacienții cu fenilcetonurie
Pentru celulele copilului nervos nu sunt expuse la efectele toxice ale fenilalanină și derivații săi, este necesar să se elimine complet din dieta de proteine animale. Apoi, creierul va fi complet sănătos, dacă o faci în primele săptămâni de viață. Și dacă vom începe să limiteze proteina la o vârstă mai târziu, este posibil să se întârzie dezvoltarea unor pauză. Dar, înapoi la sănătate a sistemului nervos și pentru a elimina modificările celulele nervoase nu vor avea succes.O dieta este necesar pentru 16-18 de ani. Aceasta este o condiție prealabilă. Este de dorit să se controleze cantitatea de proteine animale în viitor.
Dacă o femeie care are simptome de PKU au fost detectate în copilărie, intenționează să devină gravidă, ea cu siguranță trebuie să se întoarcă la dieta fără fenilalanină. Aceste restricții trebuie urmate înainte de concepție, în timpul sarcinii și alăptării.
Toate necesare pentru creșterea și dezvoltarea de aminoacizi intra în corpul de produse medicale de specialitate. De obicei, ele sunt pulbere - amestec uscat de aminoacizi. Părinții lor dau un copil bolnav, consiliere medicală și genetică liber.
Sugarii primesc un amestec special complet evacuate pe bază de lactoză hidrolizat de proteină din lapte.
Substante nutritive care trebuie să înlocuiască copiii alimente bogate in proteine naturale includ:
- peptide (enzime proteine din lapte clivate);
- aminoacizi liberi (tirozină, triptofan, cistina, histidina și taurină).
alimente dietetice speciale pentru a reface rezervele de proteine la copii de toate vârstele sunt, de asemenea, „Berlafen“, „Tsimorgan“, „Minafen“, „Aponte“.
Copiii cu PKU, puteți alăpta. Dar, în timp ce o mamă care alăptează trebuie să adere la un regim alimentar special.
In dieta de preșcolari și-vârstă școlară din meniu exclude complet produsele proteice. În lista de produse autorizate - legume, fructe, produse din amidon, uleiuri vegetale. La elaborarea meniul zilnic, trebuie să respecte cu strictețe normele de vârsta de fenilalanină.
Trebuie amintit că, pentru un corp tot mai mare de nutriție bună este vitală. Deoarece copilul zilnic necesită tirozina 120 mg per kilogram de greutate. De aceea, copiii și adolescenții cu acest diagnostic ar trebui să primească aminoacizi pentru construirea unor surse suplimentare de celule și de creștere. De asemenea, asigurați-vă că pentru a prescrie vitamine și minerale complexe. Este deosebit de important ca copilul primește norma vitaminelor C, B6 si B1, acid folic, fier, calciu și magneziu. Numărul de calorii ar trebui să fie crescut cu 30%, comparativ cu rata de zi cu zi de colegii lor.
Grupuri de produse la fenilcetonurie
Distribuit de trei grupe de produse naturale. Clasificarea se bazează pe cantitatea de fenilalanină în ele:- Lista Roșie - produsele care urmează să fie complet excluse din dieta.
- Lista Orange - sunt permise în cantități mici, sub control strict.
- Lista verde - poate fi utilizat fără restricții.
Industria produce încă două grupe de produse:
- artificiale produse low-proteine, în special pentru produse alimentare dietetice (paine, prajituri, paste)
- fructe pe bază gata piureul alimente pentru copii.
Părinții unui copil cu PKU, este important să fie în măsură să facă o dieta corect și se calculează cantitatea de fenilalanină. Pentru a face acest lucru, trebuie să aveți pe scări de mână, care fac posibil să cântărească până la o zecime de gram.
Controlul de fenilalanină din sânge
Este necesar să se controleze cantitatea de fenilalanină. Ar trebui să fie între 3-4 mg% sau 180-240 pmol / l.Pentru a determina necesitatea de a face un test de sânge în laborator. Până la vârsta de trei luni, se face o dată pe săptămână.
Treptat, medicul reduce numărul de teste. De la luni la un an - o dată pe lună, cu unul până la trei ani - o dată la două luni. După trei ani, frecvența inspecțiilor este redusă la o dată la trei luni. Există un regim special, dar un specialist poate schimba în funcție de starea pacientului.
Face analiza de preferință dimineața pe stomacul gol. Calitatea și regularitatea monitorizării și a corecțiilor în timp util în dieta depinde de păstrarea intelectului.
Răspunsuri la întrebări frecvente
Care sunt simptomele de fenilcetonurie neonatale?
Nou-născuți diagnosticați cu PKU nu diferă de la copii sănătoși. Și în cazul în care boala în timp pentru a identifica și a opri dezvoltarea sa în prezent și în viitor acest copil va fi complet sănătos.Cum sunt pacienții cu fenilcetonurie?
La nastere, copiii, pacienții cu fenilcetonurie, nu sunt diferite de alți copii. Dar, în a doua lună de viață începe să apară modificări:
- luminarea părului și a irisului din cauza lipsei de melanina
- crestere excesiva in greutate
- fontanela repede supradezvoltat
- piele dryish
- peeling, erupții cutanate și eczeme
- vărsături frecvente
- urină și sudoare de tipic miros „mouse“
- apar crampe și spasme
- rigiditate și sandwich „postura croitor“, care sunt asociate cu cresterea tensiunii musculare
- comportament inadecvat, strigând, râs
- reducerea dimensiunii craniului
- urechi deformare
- degetele tremurătoare
- incontinenta urinara
- proeminente maxilarul inferior
Ce amestec utilizat pentru copil cu fenilcetonurie?
În scopul de a oferi copiilor cu toate substanțele nutritive necesare în cantități suficiente, a dezvoltat amestecuri speciale pe bază de aminoacizi esențiali și non-esențiali. În compoziția lor include, de asemenea, vitamine și oligoelemente necesare.Pentru copiii de până la un an, se recomandă:
- Afenilak 13 Afenilak 15 de la compania "Nutritek", Rusia;
- MIDmil PKU 0 (Hero, Spania);
- XP analog ("Nutricia", Țările de Jos);
- Fenil gratuit 1 ("Mead Johnson" Statele Unite).
- AM P-1, P-2 AM, AM-R3;
- Izifen (produs finit), și HR și HR Maksameyd Maksamum arome neutre și fructe ("Nutricia", Țările de Jos).
Care este speranța de viață a pacienților cu fenilcetonurie?
În cazul în care o persoană a avut timp la tratament în consecință, durata și calitatea vieții nu a fost diferită de restul societății. În cazul în care a dezvoltat dementa, speranța de viață se reduce dramatic.Cum de a trata fenilcetonuria?
Astăzi, în Rusia folosi un regim alimentar special bezfenilalaninovuyu pentru tratamentul fenilcetonurie. Pentru realimentarea tirozină și alți aminoacizi, toți copiii bolnavi primesc o preparate speciale gratuite. Este de dorit să se mențină la o dieta de 18 ani, cu toate că unii medici spun că este mai bine să facă acest lucru pe tot parcursul vieții lor.Această metodă de terapie dieta este cel mai ieftin si eficient. De-a lungul anilor, el îi ajută pe copiii cu aceasta boala sa creasca sanatosi. În dezvoltarea sa, acestea sunt în nici un fel inferioare colegii lor. Cei care au un copil diagnosticat „fenilcetonurie“ în școală, absolvent, începe o familie și de a da naștere la copii sănătoși.
Rezumând, observăm că este greu de fenilcetonuria este o tulburare genetica care poate duce la handicap mintal. Modificările apar în sistemul nervos foarte repede si sunt ireversibile. Cu toate acestea, puteți preveni dezvoltarea bolii. Acest lucru necesită diagnostic precoce și un regim alimentar special.
Care sunt tipurile de fenilcetonurie?
Izolați 3 tipuri de fenilcetonurie:- fenilcetonurie eu. Forma clasică și cea mai comună a bolii, așa cum este descris mai sus in articol. Acesta este asociat cu o mutatie intr-o gena pe cromozomul 12, în care formarea este perturbat enzimatic fenilalanină-4-hidroxilazei, care convertește fenilalanina la tirozina.
- fenilcetonurie II. În această formă a bolii apare în încălcarea cromozomul 4-a. Producția este enzimă deranjat digidropteridinreduktazy, care promovează, de asemenea, conversia de fenilalanină la tirozina. Boala este moștenită în același mod ca am forma: a fost născut un copil bolnav, este necesar să se efectueze gena au ambii părinți. Prevalența fenilcetonuriei II - 1 caz la 100 000 născuți vii.
- fenilcetonurie III. Ca urmare, există o lipsă de enzimă tulburări genetice 6 piruvoiltetragidropterinsintazy. Heritage, precum și cele două forme anterioare ale bolii. Prevalența - 1 la 300.000 de nașteri.
fie de invaliditate la fenilcetonurie da?
Criteriile de invaliditate în fenilcetonurie:- Când fenilcetonurie I de invaliditate ajustate numai atunci când tulburări ireversibile ale sistemului nervos central, care duc la tulburari neurologice si retard mental.
- Când fenilcetonuria II și tip III grup de invaliditate stabilit în toate cazurile.
Există prevenirea fenilcetonurie?
fenilcetonurie speciale de prevenire nu există. Dar unele evenimente de ajutor pentru a evalua corect riscurile, în timp pentru a lua măsurile necesare:- consiliere genetică. Este necesar ca persoanele care intenționează să aibă copii care sunt bolnavi sau sunt purtători ai genei greșit, care este bolnav cel puțin o rudă apropiată sau se naște un copil bolnav. Consilierea conduce medic-genetician. Aceasta ajută să înțelegem modul în care gena responsabilă de fenilcetonurie, a trecut în generațiile anterioare, care sunt riscurile viitorului copil. De asemenea, genetician ajută la planificarea familiei.
- screening-ul nou-nascuti. Analiza nu ajuta la prevenirea bolii, dar o identifică mai devreme, atâta timp cât nu a condus încă la schimbări ireversibile la nivelul creierului.
- Consultare si dieta pentru femeile care suferă de fenilcetonurie. Dacă sunteți o femeie și suferă de fenilcetonurie, ar trebui să se consulte cu medicul dumneavoastră și întrebați atunci cel mai bine este de a planifica sarcina în cazul dumneavoastră. In timpul sarcinii, trebuie să urmați dieta dreapta - ajută la prevenirea dezvoltarea defectelor copilului.
Care este prognoza pentru fenilcetonurie?
Prognosticul depinde de forma de boală și tratament precoce, respectarea recomandărilor dietetice, de corecție medicale și educaționale.Când fenilcetonurie I, la începutul măsurilor necesare în timp util, prognosticul este favorabil. Un copil creste si se dezvolta in mod normal. Dacă ți-e dor de un tratament, si dieta, rezultatul nu este atât de bun.
Când fenilcetonuria II și III prognostic grav. Dietele nu face efect.
Care sunt factorii de risc pentru fenilcetonurie?
- După cum sa menționat în articol, copilul prezintă riscul de a avea boala sau sa fie un purtător al genei mutante, în cazul în care există în ambii părinți.
- Printre diferitele grupuri etnice în prevalența fenilcetonurie variază. De exemplu, gena anormala este mai putin frecvente printre negri.
- În grupul cu risc ridicat sunt copiii mamelor care suferă de fenilcetonurie. Dacă în timpul sarcinii femeia nu urmeze o dietă specială, copilul poate avea defecte de dezvoltare.
Sfaturi pentru pacienții cu fenilcetonurie
- În mod constant monitorizată cu atenție. Dacă dumneavoastră sau copilul dumneavoastră trebuie să rămânem la o dieta saraca in fenilalanina, trebuie să țină o evidență a produselor alimentare de zi cu zi consumate.
- Încercați să păstrați calculele au fost cât mai exacte posibil. Utilizați special ceașcă de măsurare, lingura, scale care poate măsura greutatea în grame. Acesta va monitoriza îndeaproape cantitatea de fenilalanină consumate în fiecare zi.
- Utilizați pentru a înregistra cantitatea de fenilalanină în dieta dumneavoastră sau în jurnal dieta alimentară copilului dumneavoastră sau un program special de calculator.
- Nu neapărat mult mai limitati. Astăzi puteți cumpăra alimente speciale, sărac în fenilalanină, cum ar fi paste, orez, făină, pâine, și să mănânce aproape ca o persoană obișnuită.
- Fii creativ. Discutați cu medicul dumneavoastră, dietetician: poate că te va sfătui cum puteți diversifica dieta fără a sacrifica sănătatea. Puteți face mese mai diversă prin condimente.
 În cazul în care este necesară galactosemie, deoarece este posibil să se efectueze un studiu în…
În cazul în care este necesară galactosemie, deoarece este posibil să se efectueze un studiu în… Atunci când ereditatea obraznic: semne fenilcetonuriei și căile de atac de ajutor populare
Atunci când ereditatea obraznic: semne fenilcetonuriei și căile de atac de ajutor populare Ce este fenilcetonuria?
Ce este fenilcetonuria? Am schimbat mirosul de urină: tipurile de mirosuri și posibile boli
Am schimbat mirosul de urină: tipurile de mirosuri și posibile boli Microcefalie. Cauze, simptome, patologie, diagnostic
Microcefalie. Cauze, simptome, patologie, diagnostic Îndulcitorii în diabet
Îndulcitorii în diabet Toate de comprimate efervescente Supradin: comentarii
Toate de comprimate efervescente Supradin: comentarii Tabletele și pastile pentru supt laringită
Tabletele și pastile pentru supt laringită Gaviscon - descriere și instrucțiuni de aplicare
Gaviscon - descriere și instrucțiuni de aplicare Manualul de instrucțiuni Ambroxolul
Manualul de instrucțiuni Ambroxolul Spirulina: proprietățile sale utile și contraindicații
Spirulina: proprietățile sale utile și contraindicații Aspartamul - avantaje și prejudicii sale, utilizarea
Aspartamul - avantaje și prejudicii sale, utilizarea De droguri in timpul menopauzei Estrovel 1
De droguri in timpul menopauzei Estrovel 1 Ekstravel de droguri în menopauză
Ekstravel de droguri în menopauză Opinii de colagen ultra
Opinii de colagen ultra Accident vascular cerebral hemoragic: efecte, cauze, reabilitare și prognoză
Accident vascular cerebral hemoragic: efecte, cauze, reabilitare și prognoză Cauzele miros urât de urină la copii, adulți și sarcina
Cauzele miros urât de urină la copii, adulți și sarcina Ciulin Grass: proprietăți și aplicații utile
Ciulin Grass: proprietăți și aplicații utile Phenylketonuria se caracterizeaza prin alterarea metabolismului
Phenylketonuria se caracterizeaza prin alterarea metabolismului Ekstravel de droguri în menopauză
Ekstravel de droguri în menopauză Accident vascular cerebral hemoragic: efecte, cauze, reabilitare și prognoză
Accident vascular cerebral hemoragic: efecte, cauze, reabilitare și prognoză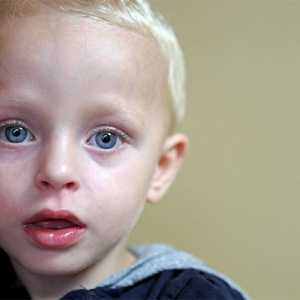 În cazul în care este necesară galactosemie, deoarece este posibil să se efectueze un studiu în…
În cazul în care este necesară galactosemie, deoarece este posibil să se efectueze un studiu în… Toate de comprimate efervescente Supradin: comentarii
Toate de comprimate efervescente Supradin: comentarii Spirulina: proprietățile sale utile și contraindicații
Spirulina: proprietățile sale utile și contraindicații Ce este fenilcetonuria?
Ce este fenilcetonuria? Îndulcitorii în diabet
Îndulcitorii în diabet Atunci când ereditatea obraznic: semne fenilcetonuriei și căile de atac de ajutor populare
Atunci când ereditatea obraznic: semne fenilcetonuriei și căile de atac de ajutor populare